Fusarium root rot, produced by Fusarium solani f. sp. pisi
is an important disease of pea in many regions of the world (4, 6).
Complete resistance to this disease has not been reported in pea, but a
number of sources of partial tolerance have been found (4).
Recently, Gr�nwald et al. (2) screened the Pisum Core Collection and
identified 44 accessions with some level of tolerance (mean disease severity
rating of 2.5 or less on a scale of 0 to 5).
Most of these accessions were purple-flowered P. sativum ssp. sativum
lines, a number of which originated from
Afghanistan
. None of the tolerant lines were
identified as being P. s. ssp. elatius or P. s. ssp. abyssinicum.
We were therefore surprised to find that in a small screen of lines we
have used as parents in mapping populations, a P. s. ssp. elatius
line (JI 1794) exhibited good tolerance to this pathogen.
Here we present the results of an analysis designed to locate the primary
genetic factors responsible for the increased tolerance in JI 1794.
Materials
and Methods
The population used for this study was the recombinant inbred line (RIL)
population derived from a cross between JI 1794 (Pisum
sativum ssp. elatius var. pumilo)
and Slow, (Pisum sativum ssp. sativum).
This population consists of 51 F2-derived F10+
lines with excellent marker coverage for genetic mapping.
It has been used to form the basis of the consensus map of pea (11).
A standard susceptible control line (Dark Skin Perfection) and an
accession from Afghanistan known to possess a good level of tolerance (PI 223285) were also used in the
analysis.
The cultures of Fusarium
solani used in this experiment are referred to as strain 915 and were
isolated from the pea trial fields on Spillman Farm in Pullman, WA
with the assistance of K. McPhee and D. Mathre.
The soil used for the inoculum
was a 3:1 river sand and silt loam mixture that had been sterilized.
Inoculum soil was prepared according to a method previously
described (5), except that Czapek-Dox broth was used in place of Kerr�s liquid
medium, and the inoculated soil was mixed by hand.
Pasteurized soil mix, consisting of equal parts silt loam, river sand,
and Sunshine peat moss, was obtained from the Plant Growth Center at Montana State
University. The soil was placed in ten flats
to a depth of 5 cm. Five seeds for
each pea line (a susceptible control [Dark Green Perfection], a tolerant control
from Afghanistan [PI223285], the two parents and 51 RILs) were collected and surface sterilized
for five minutes in 10% chlorine bleach. The
seed coats were then nicked and placed between moistened filter paper and
allowed to imbibe for 12 hrs. The
seeds were placed on the soil surface in the flats (in a predetermined
randomized pattern), and 2 grams of inoculum were placed on top of each seed.
An additional 2 cm layer of soil mix was added over the inoculated seeds,
and the flats were thoroughly watered. Flats
were watered to bring soil to field capacity.
Soil moisture was maintained between field capacity and leaf wilting
point in order to avoid over saturation and the development of damping off
diseases such as Pythium. Greenhouse
conditions provided 14 hours of light per day, four of which (two in the morning
and two in the afternoon) were supplemented by growth lights.
Disease incidence was determined using two methods: a disease score and a
tolerance score.
Disease
score:
Plants were carefully extracted from the soil after four weeks, and the
roots were thoroughly rinsed. Symptoms
of discoloration and disfiguration of the roots and epicotyl were scored
visually on a scale of 0 to 5 (3), with 0 indicating negligible evidence of
disease and 5 indicating complete rot, as shown in Fig. 1.
Within most lines the disease score was relatively consistent (+1
unit on our scale). An average score
for each line was calculated and used for analysis and genetic mapping.
 Tolerance
score:
During the analysis a number of plants were observed that despite a high
disease score displayed vigorous vegetative growth, and we were uncertain
whether we were capturing all the disease effects by simply using the
traditional disease score. We
therefore developed a second scoring method we designate the �tolerance
score� in which the vigor of the vegetative growth is also taken in to
account. The tolerance score
differed from the disease score in that the original score given to each plant
was increased, decreased, or left the same depending on the correlation (or lack
thereof) of the above ground symptoms to the expected above ground symptoms
based on the disease score. For
example, for a plant with a disease score of 4, the above ground portion would
be expected to be wilted or drying out. If
that were the observed case, the score of 4 would be retained for the tolerance
score. If the above ground portion
of the plant appeared healthy (a condition expected for a disease score of 2),
then the tolerance score would be changed to a 3, thereby averaging the scores
for observed and expected symptoms.
Tolerance
score:
During the analysis a number of plants were observed that despite a high
disease score displayed vigorous vegetative growth, and we were uncertain
whether we were capturing all the disease effects by simply using the
traditional disease score. We
therefore developed a second scoring method we designate the �tolerance
score� in which the vigor of the vegetative growth is also taken in to
account. The tolerance score
differed from the disease score in that the original score given to each plant
was increased, decreased, or left the same depending on the correlation (or lack
thereof) of the above ground symptoms to the expected above ground symptoms
based on the disease score. For
example, for a plant with a disease score of 4, the above ground portion would
be expected to be wilted or drying out. If
that were the observed case, the score of 4 would be retained for the tolerance
score. If the above ground portion
of the plant appeared healthy (a condition expected for a disease score of 2),
then the tolerance score would be changed to a 3, thereby averaging the scores
for observed and expected symptoms.
The disease incidence data were analyzed in two ways.
The ten most tolerant lines and the ten most susceptible lines were used
as the tails of the distribution and were analyzed for correlation with
segregating marker loci using QUIKMAP (10).
For each locus that displayed significant skewing (7 or more tolerant
lines with the allele from one parent and 7 or more susceptible lines possessing
the allele from the other parent) the entire population was divided into two
groups based on genotype at that locus. An
average tolerance score for each group was calculated and the difference tested
for significance using a two-tailed Student�s t-test.
The second approach was a traditional analysis for quantitative trait
loci using QTL Cartographer (9) and a set of nearly 200 loci spaced about 4 cM
apart along each of the linkage groups.
Results
Most lines germinated well, and the plants appeared healthy above ground
throughout the experiment. However,
four lines displayed poor germination. Two
of these lines lacked overall vigor even when growing in clean soil, and the
poor germination could be attributed to poor seed quality.
However, the other two lines gave excellent germination and emergence
when placed in pasteurized soil. Because
root symptoms could not be adequately assessed for these lines, we did not
include the two �low vigor� lines in our final analyses and performed the
two QTL analyses both with and without data from the other two lines (the few
plants from these lines that did emerge displayed a highly susceptible phenotype
of 4 or 5).
For each plant, disease symptoms were scored as described above.
Average disease scores ranged from 1.10 to 4.85, with JI 1794 scoring
1.25 and Slow scoring 3.30. The
susceptible control gave an average disease score of 3.90 and PI223285 gave an
average score (1.35) very similar to JI 1794.
Average tolerance scores ranged from 1.10 to 4.85, with JI 1794 scoring
1.06 and Slow scoring 2.85. Scores
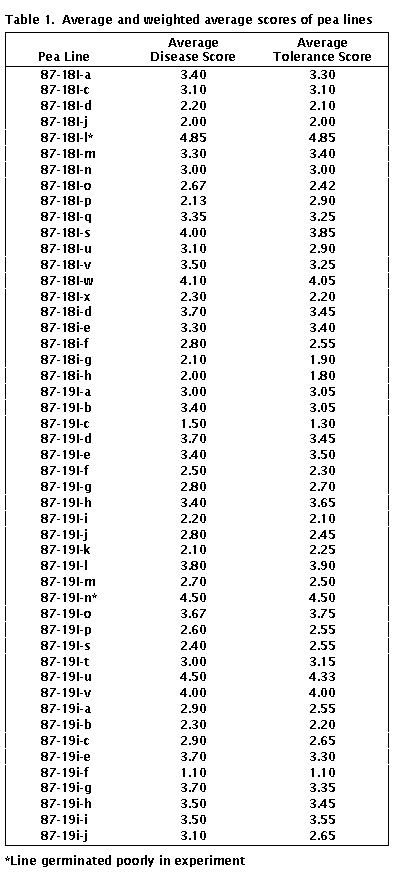
for individual RILs are listed in Table 1.
The QUIKMAP analysis revealed four potential regions on the linkage map
correlating with the fluctuation in susceptibility.
These regions included the portion of LG III around M, a portion
of LG III near Le, a section of LG IV just proximal to the ribosomal
array and the region on LG VI distal to Gty.
Only the last of these regions showed a significant difference between
the averages for the two alleles using Student�s t-test (average disease score
for lines with JI 1794 allele = 2.75 and for lines with the Slow allele = 3.29;
P = 0.02). Values for the average
tolerance scores were similar (JI 1794 = 2.69, Slow allele = 3.20; P = 0.03).
The region on LG IV showed a P = 0.08 for both the average disease score
and the average tolerance score, while the other two regions had P > 0.10 for
both averages. The two-locus average
genotypic scores (j = JI 1794 allele and
s = Slow allele with the locus on
LG VI given first) are as follows jj = 2.44,
js = 3.08, sj = 3.29 and ss =
3.29, indicating that the locus on LG VI had the major effect, with the j allele
on LG IV providing additional tolerance in the presence of the j allele on LG
VI.
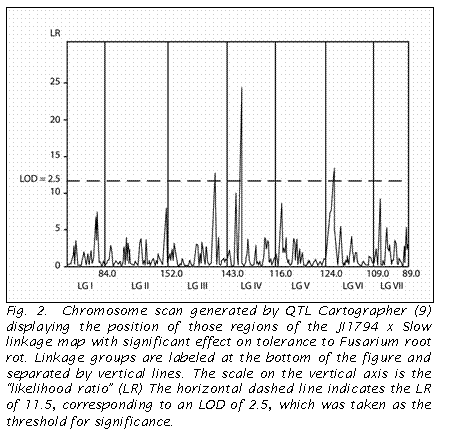
 The analysis using QTL Cartographer identified three QTLs with an LR of
11.5 (= LOD 2.5) or greater (Fig 2). These
three QTLs corresponded to the regions on LG VI and LG IV revealed in the
QUIKMAP analysis, as well as that near Le on LG III.
In contrast to the QUIKMAP analysis, QTL Cartographer indicated a greater
effect by the region on LG IV.
The analysis using QTL Cartographer identified three QTLs with an LR of
11.5 (= LOD 2.5) or greater (Fig 2). These
three QTLs corresponded to the regions on LG VI and LG IV revealed in the
QUIKMAP analysis, as well as that near Le on LG III.
In contrast to the QUIKMAP analysis, QTL Cartographer indicated a greater
effect by the region on LG IV.
Discussion
The susceptibility segregation patterns observed in this study, and the
analyses thereof, indicate that tolerance to F. solani is multigenic in
JI 1794. Both methods of assessing
susceptibility (disease score and tolerance score) identified the same regions
of the genome as influencing the trait. Because
the former method is traditional and less subjective, we recommend the continued
use of the disease score as the standard analysis.
The QUIKMAP analysis and QTL Cartographer also gave very similar results,
although QUIKMAP revealed only one region with a significant effect.
The effects of the regions on LG IV and LG VI are particularly strong,
and we are currently introgressing these regions into a more acceptable genetic
background for commercial use.
The position on the linkage map of the factors influencing tolerance in
this study may provide some indication as to what these genes might be.
There are several genes influencing susceptibility to fungal diseases on
LG VI (1, 7, 8). However, at least Er1
and the QTL identified for tolerance to Aphanomyces root rot are located on the
opposite side of Gty from the QTL for Fusarium root rot.
Indeed, the position of Er1 has been accurately determined in the same JI 1794
x Slow
RIL population used in this study, and several recombinant lines can be
identified. Similarly, the position
of the Fusarium root rot QTL on LG IV is nearly 50 cM from the Aphanomyces root
rot QTL reported on this same chromosome (12).
JI 1794 is known to have smaller roots than Slow, and a major gene
influencing this trait is located very near Le (13).
The smaller root mass of JI 1794 could, in some way, be responsible for
the QTL for Fusarium root rot tolerance identified in this study to be near Le.
Other than this possible connection between Le and the minor QTL,
there do not appear to be any obvious candidate genes that could explain the
tolerance found in JI 1794.
The
primary source of Fusarium root rot tolerance reported in pea has been
accessions from Afghanistan (2). The genetic factors
responsible for this tolerance have not been analyzed extensively, and we do not
know if the genes found in JI 1794 are different from those present in the Afghanistan
material. An analysis of allozyme
diversity in pea (14) indicated that the Afghanistan material formed a unique subset of the Pisum sativum ssp. sativum
germplasm, not closely related to P. s. ssp. elatius.
Hence, Fusarium root rot tolerance either arose independently in the two
lineages or dates back to a very early Pisum sativum form and has
subsequently been lost in most other pea lines.
1. Cargnoni, T.L.,
Weeden, N.F. and Gritton, E.T. 1994.
Pisum Genetics 26: 11-12.
2. Gr�nwald, N.J., Coffman, V.A. and Kraft, J.M. 2003.
Plant Dis. 87: 1197-1200.
3. Kraft, J.M.
1989. Pisum Newslett. 21:
82-85.
4. Kraft, J.M., Haware,
M.P., and Hussein, M.M. 1988.
In: Summerfield, R.J. (ed.) World Crops: Cool Season Food Legumes.
Kluwer Academic Press, Boston. pp 565-575.
5. Kraft, J.M., and
Roberts, D.D. 1969.
Phytopath. 59: 149-152.
6. Kraft, J.M. and
Pfleger, F.L. 2001.
Compendium of Pea Diseases and Pests. Second Ed.
Amer. Phytopath. Soc., St. Paul, MN, 67 pp.
7. Pilet-Nayel, M.L.,
Muehlbauer, F.J., McGee, R.J., Kraft, J.M., Baranger, A., and Coyne, C.J. 2002.
Theor. Appl. Genet. 106: 28-39.
8.
Timmerman, G.M., Frew, T.J., Weeden, N.F., Miller, A.L. and Goulden, D.S.
1994. Theor. Appl. Genet. 88:
1050-1055.
9. Wang, S. and Zeng, Z-B.
2003. Windows QTL
Cartographer v2.0. Bioinformatics Research Center. North Carolina State Univ.
10.
Weeden, N.F. and Barnard, J. 1994.
NYSAES, Geneva, NY. Computer Ctr. Tech. Rpt. 20-2.
11.
Weeden, N.F., Ellis, T.H.N., Timmerman-Vaughan, G.M., Święcicki, W.K., Rozov, S.M. and Berdnikov, V.A.
1998. Pisum Genetics 30: 1-4.
12.
Weeden, N.F., McGee, R., Grau, C.R. and Muehlbauer, F.J.
2000. Pisum Genetics 32:
53-55.
13.
Weeden, N.F. and Moffet, M. 2002.
Pisum Genetics 34: 28-31.
14.
Weeden, N.F. and Wolko, B. 1988.
Measurement of genetic diversity in pea accessions collected near the
center of origin of domesticated pea. IPBGR
final report, Rome, 20 pp.